West syndrome (WS) is a devastating pathological condition occurring in circa 1 every 3,500 infants. It includes infantile-onset spasms, prominent interictal EEG abnormalities and impaired psychomotor development. Spasms and EEG abnormalities often disappear before 3 years of age. However, the majority of patients will develop other types of epileptic syndromes. WS etiopathogenesis is extremely complex. A subset of WS cases is linked to defined gene lesions. In particular, in up to 5% of cases, WS has been associated to 14q12 microduplications including the transcription factor gene FOXG1.
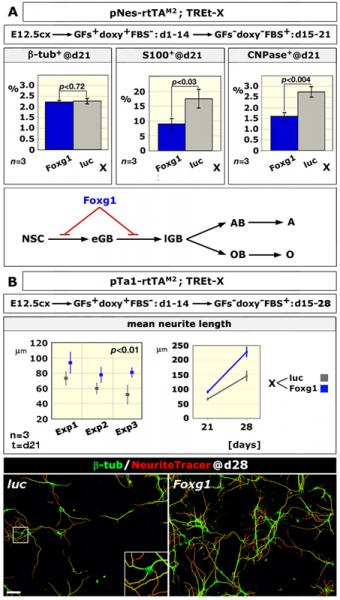 We discovered that Foxg1 overexpression in murine cortico-cerebral precursors alters the astrocyte-to-neuron generation ratio and induces a prominent neuritic overgrowth, two phenomena likely to contribute to WS pathogenesis. Moreover, we suspect that an alteration of the cortical GABAergic complement, stemming from early mispatterning of telencephalon, might further concur to the profile of WS patients. Now we want to thoroughly verify the contribution of these phenomena to WS etiopathogenesis.
We discovered that Foxg1 overexpression in murine cortico-cerebral precursors alters the astrocyte-to-neuron generation ratio and induces a prominent neuritic overgrowth, two phenomena likely to contribute to WS pathogenesis. Moreover, we suspect that an alteration of the cortical GABAergic complement, stemming from early mispatterning of telencephalon, might further concur to the profile of WS patients. Now we want to thoroughly verify the contribution of these phenomena to WS etiopathogenesis.
For this purpose, we developed mouse models conditional gain-of-function for Foxg1 and we are differentiating patient-specific induced pluripotent cells (iPSCs) to pallial- and subpallial-like neural tissue. Mutant mice will be tested with the help of our Collaborators, for their capability to reproduce key features of WS patients, via behaviourial analysis, electroencephalo-graphy and in situ expression profiling of immediate-early genes. Both mutant mice and patient-specific cortical tissue will be subject of neuroembryological studies. Finally, as a specificity control, we will assay if RNAi-correction of FOXG1 expression levels antagonizes possible histogenetic and functional abnormalities displayed by patient iPSC-derived neural precursors.